Chemissian news
[27 May 2023] Chemissian v4.80 released
- NEW Added: Chemissian can now calculate spin natural orbitals as the eigenvectors of the spin density matrix in UHF/UDFT calculations. This new feature is valuable for analyzing the nature of unpaired electrons.
For a given UHF/UDFT calculation click the "a-b" button in the Structure window. Chemissian will calculate the engenvectors of the spin density matrix. One can analyze and plot these spin natural orbitals using:
- Tools -> Analyze MO compositions
- Tools -> Analyze electronic density
- Improved support of Gaussian output files
- Other small improvements.
[5 May 2020] Chemissian v4.67 released
- Improved support of NWChem and Gaussian output files.
- Other small improvements.
[26 August 2018] Chemissian v4.60 released
- Improved support of ORCA output files with TDDFT/CIS spectra.
- Other small improvements.
[16 July 2017] Chemissian v4.52 released
- Fixed bug with reading Gaussian output files.
[5 July 2017] Chemissian v4.51 released
- NEW option to save 3D images in good quality.
Use the "Analyze electron density" tool to plot the 3D surface, then click Plot->Export and select "High resolution Bmp file".
Your picture will be saved in a high quality as a Bmp file.
- Improved support of Spartan, Gamess, Turbomole-MOLDEN and Gaussian output files.
- Other small improvements.
- Notes to Spartan users have been updated (see here)
[17 May 2016] Chemissian v4.43 released
- Improved support of Gaussian output files.
- Fixed bug with reading NWChem transitions.
- Other small improvements.
[29 April 2015] Chemissian v4.38 released
- Improved support of Gaussian output files.
- Considerably increased speed of plotting MO diagrams with contributions (see here).
- Fixed bug with plotting beta-> beta natural transition orbitals.
- Other small improvements.
[26 February 2015] Chemissian v4.36 released
- NEW Added support of geometry optimization output files generated by Gaussian.
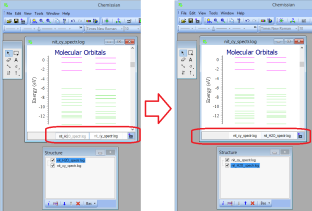
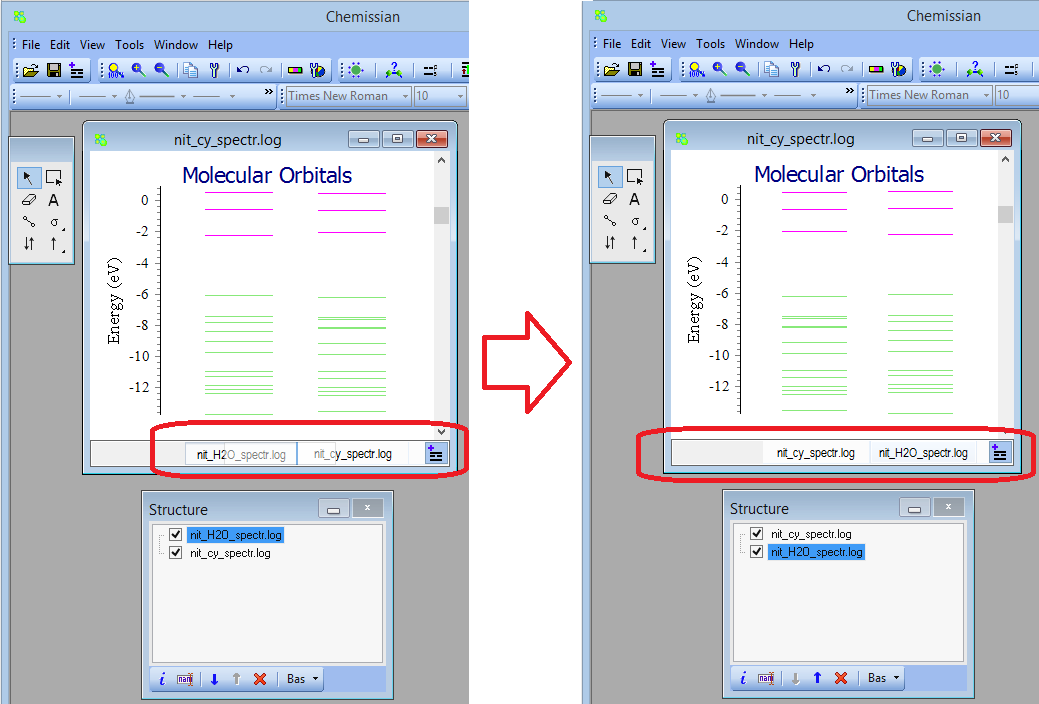
Generally, Chemissian supports only single point calculations - NEW If there are several MO diagrams it is possible to exchange the diagrams by "drag and drop" (you can also do it in the structure window)
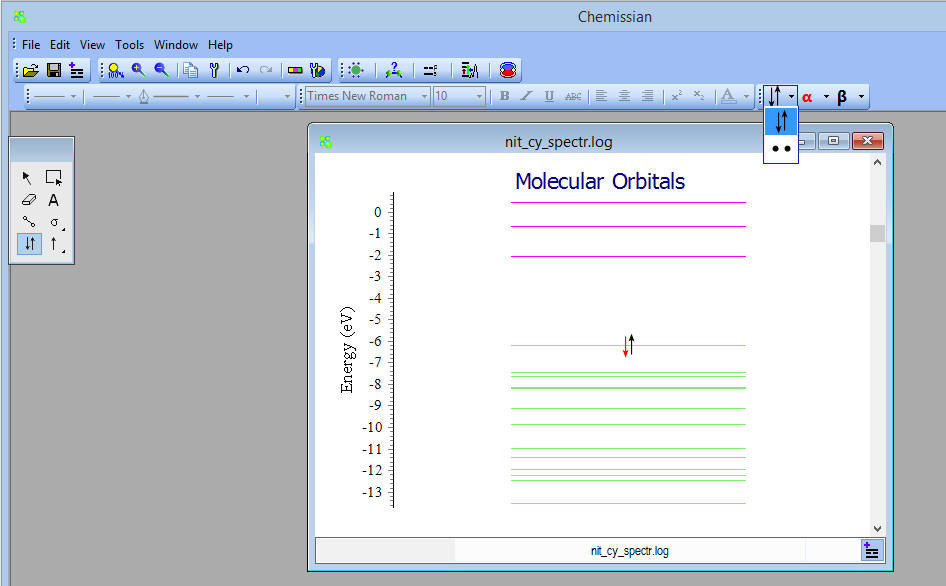
- NEW Added possibility to customize electrons:
- Other small improvements.
[30 October 2014] Chemissian v4.33 released
- NEW Added numerous options for customization of 3D.
- Fixed bugs with reading some kink of Spartan2014 files
- Now Peaks/Shapes of electronic spectra can be exported in nm/eV/a.u./cm-1
- Improved support of EOM-CCSD spectra from NWChem output files
- Now one can see percentage contributions from MO transitions.
For this check/uncheck "View->Show percentage contributions" menu item in the "Analyze spectrum" window. - Other small improvements.
[6 June 2014] Chemissian v4.23 released
- Fixed bug with calculation and vizualization of NTOs in some cases (when the number of virtual orbitals is less than the number of occupied orbitals)
[15 May 2014] Chemissian v4.2 released
- NEW Added support of EOM-CCSD spectra from NWChem output files
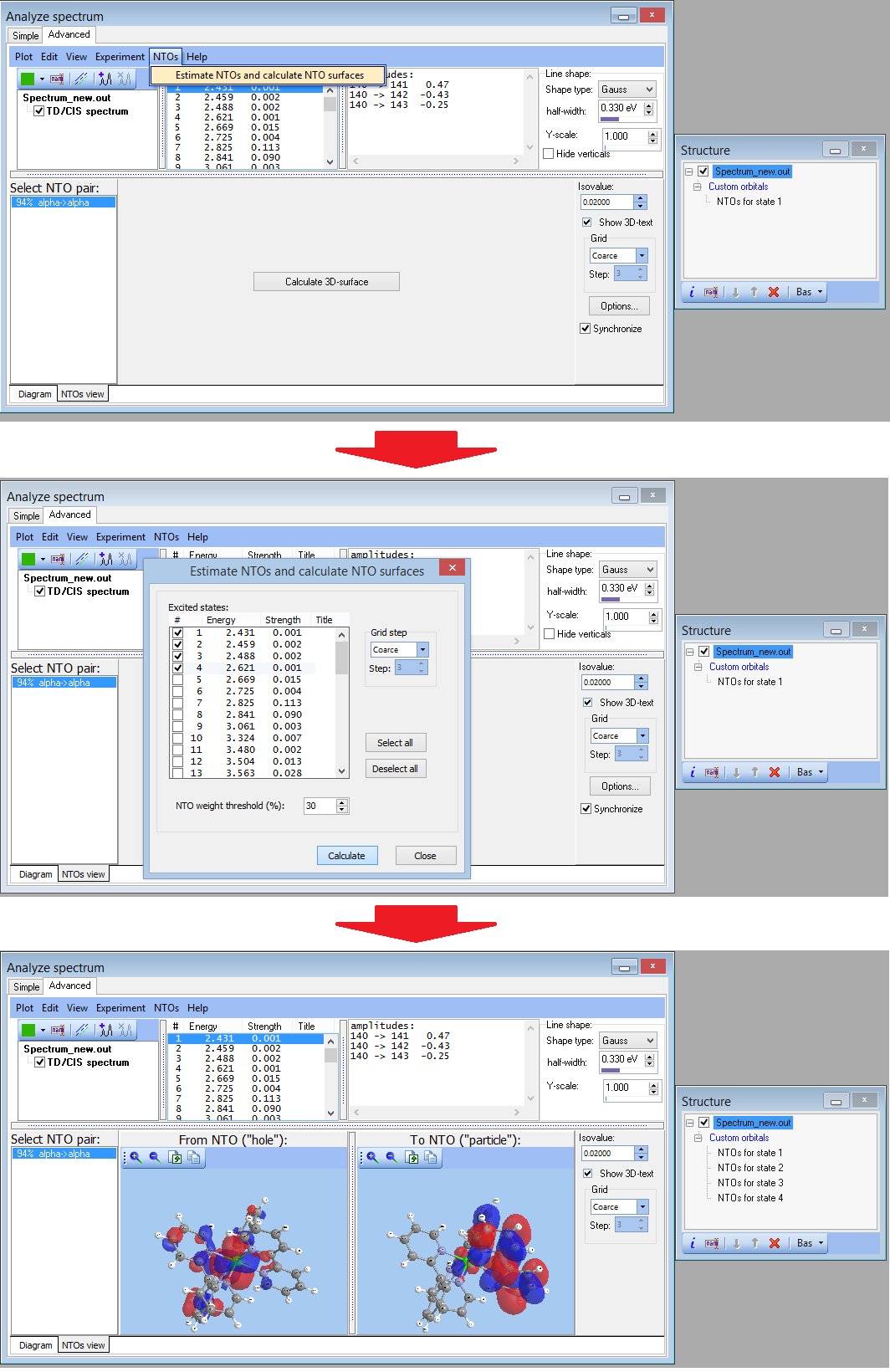
- NEW Visualization and estimation of natural transition orbitals for EOM-CC
- NEW Shape shift option:
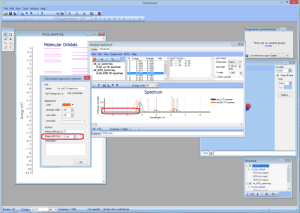
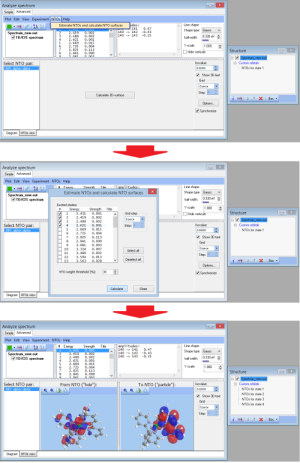
- NEW Batch feature for calculation NTOs:
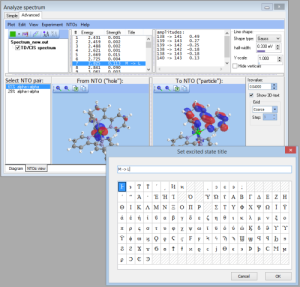
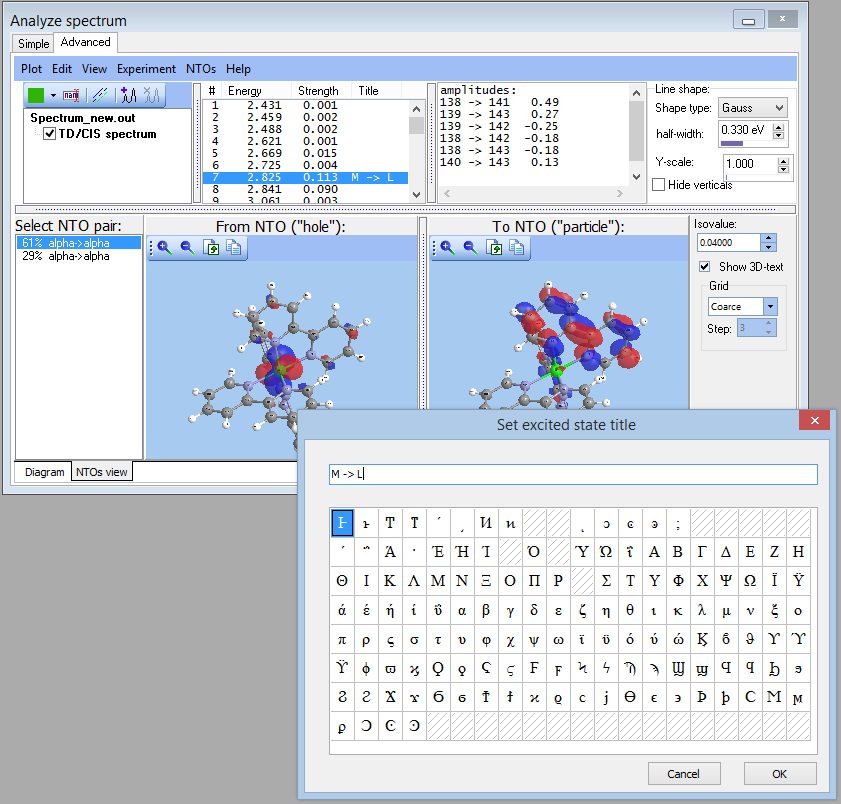
- NEWTransition title:
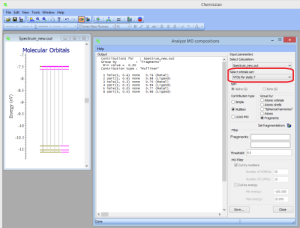
- NEWAnalysis of composition of natural trantition orbitals in terms of atomic orbitals, molecular fragments, etc. via Tools->Analyze MO compositions:
- Test support of Gaussian ONIOM output files (see Notes to Gaussian users)
- Other small improvements
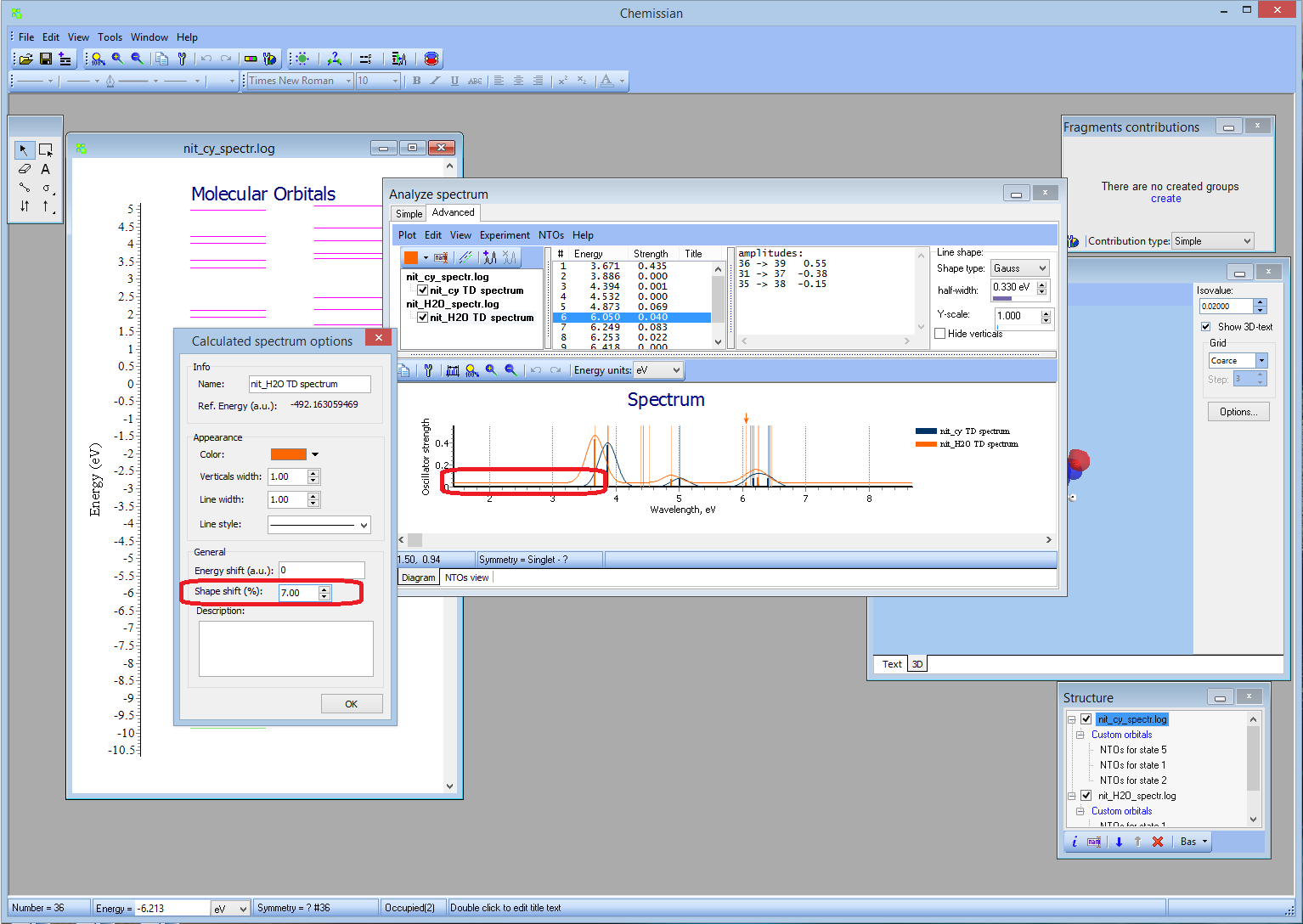

[21 January 2014] Chemissian v4.0 released
- NEW Visualization and computation of natural transition orbitals:
- Partial support of Turbomole and ORCA files via MOLDEN format
- Possibility to save MO diagram as "High resolution image".
- New options for energy axis (MO diagram)
- Support of NWChem 6.3
- Support of Windows® 8 and Windows® 8.1
- Other small improvements
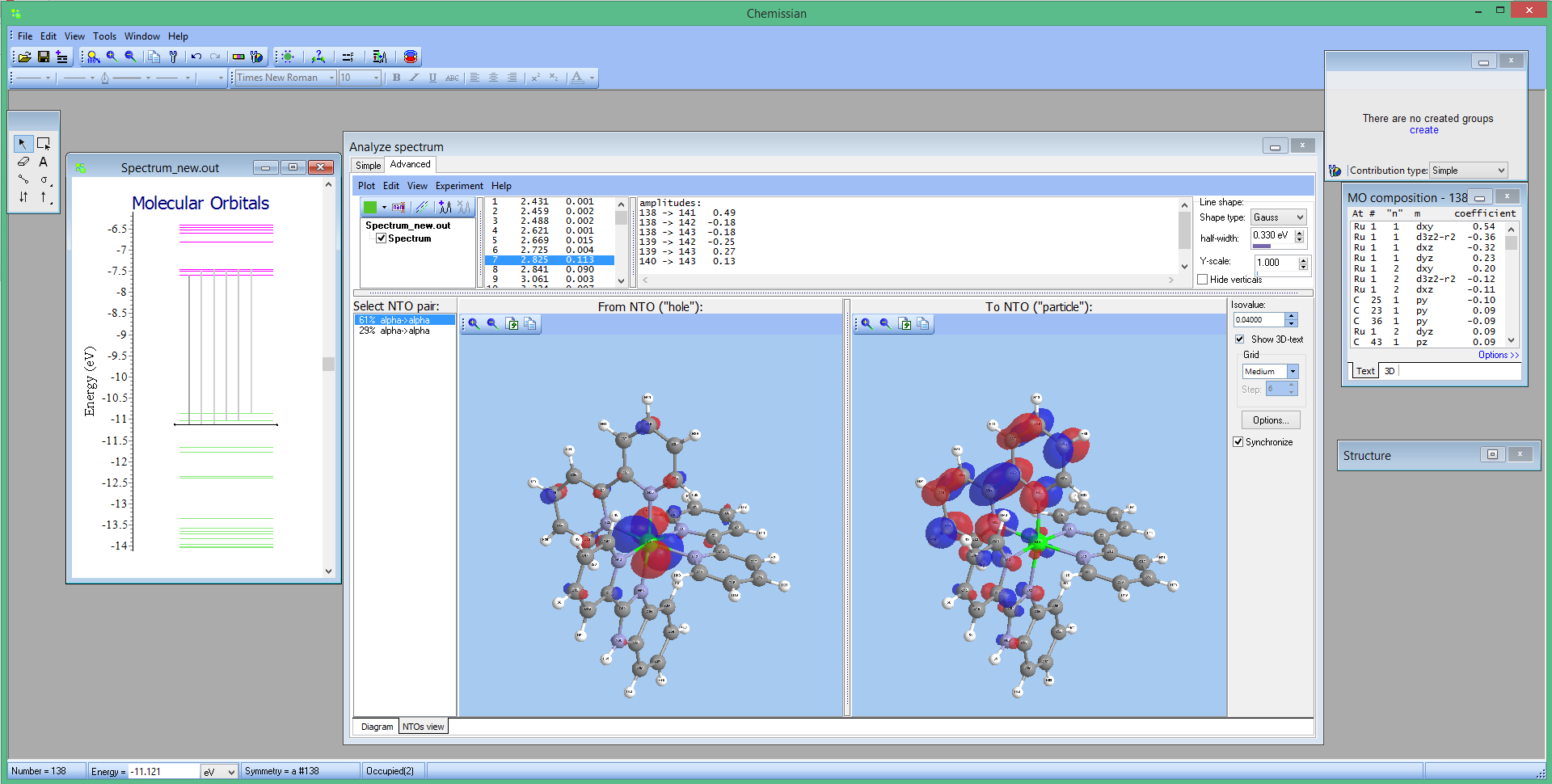
[08 September 2012] Chemissian v3.3 released
- NEW Visualisation of TDDFT UV-VIS spectra from NWChem output files
- Considerably increased speed of 3D-surfaces rendering
- Improvements in the Plot & Analyze UV-VIS spectrum Tool:
- The spectra diagram is now scaled automatically to fill all the window (by default). However, it is still possible to manually set height and width in the spectrum options window (Edit -> Edit spectrum options).
- Export line spectrum to a text file (Plot -> Export calculated peaks)
- Export spectrum shape to a text file (Plot -> Export calculated shapes).
In an exported *.txt file there will be two columns.
The first column corresponds to the energy values (wavelengths) (in selected units).
The second is the shape-function value.
If there are several calculated spectra there will be additional columns. - Energy-axis in , with variable increments. You can manually set increments for supported energy units (eV, nm, cm-1, a.u.). For this go to Edit -> Edit spectrum options > Energy axis (or just double click somewhere near the bottom axis). Note: there is also "Labels antioverlap" checkbox. Checking it will prevent overlapping of labels at the energy axis.
- Possiblily to specify input energy unit for experimental spectrum.
- Fixed some bugs with scaling experimental spectrum.
- Support of large (> 2 GB) NWChem-output files.
To use NWChem-ouput files it is necessery to run NWChem-job with print "final evals" "final vectors" option (see Notes to NWChem users).
[14 July 2012] Chemissian v3.25 released
- NEW
Export basis and geometry to NWChem input file (see structure window):
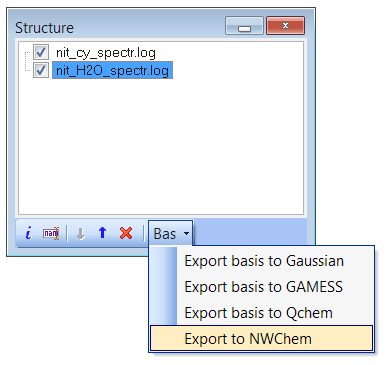
- Fixed bug with the wrong assignment of norm-factors and phases to NWChem atomic orbitals
Fixed bug regarding reading some kind of Firefly output files
[7 July 2012] Chemissian v3.2 released
- NEW
Initial support of NWChem output files
Currently, only NWChem v.6.1.1 output files were tested.
Basic support includes: 1D/2D/3D-vizualization of MOs, densities, spin densities, etc.; Calculation of population and valences; Calculation of bond orders; Calculation of MO compositions; etc.
To produce "correct NWChem output" use the "print debug" keyword in the scf section of *.nw-input.
- Fixed bug regarding reading some kind of Molpro output files (in molden format).
[1 July 2012] Chemissian v3.1 released
- Now population analysis is possible even if MOs are linearly dependent.
- Fixed some bugs regarding converting LCAO-MO to "pure" form.
[June 2012] Chemissian v3.0 released
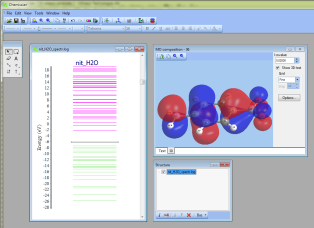
- NEW 3D
surfaces:
- At the bottom of the "MO compositions" window, there is now a small button
"3D".
Thus, it is possible now to click at a molecular orbitals energy level at the MO energy diagram and immediately see 3D-surface of this MO:
- More complex surfaces (such as spin density or some linear compilation on densities) can be plotted in "Tools --> Analyze electron density" at the "3D" tab
- At the bottom of the "MO compositions" window, there is now a small button
"3D".
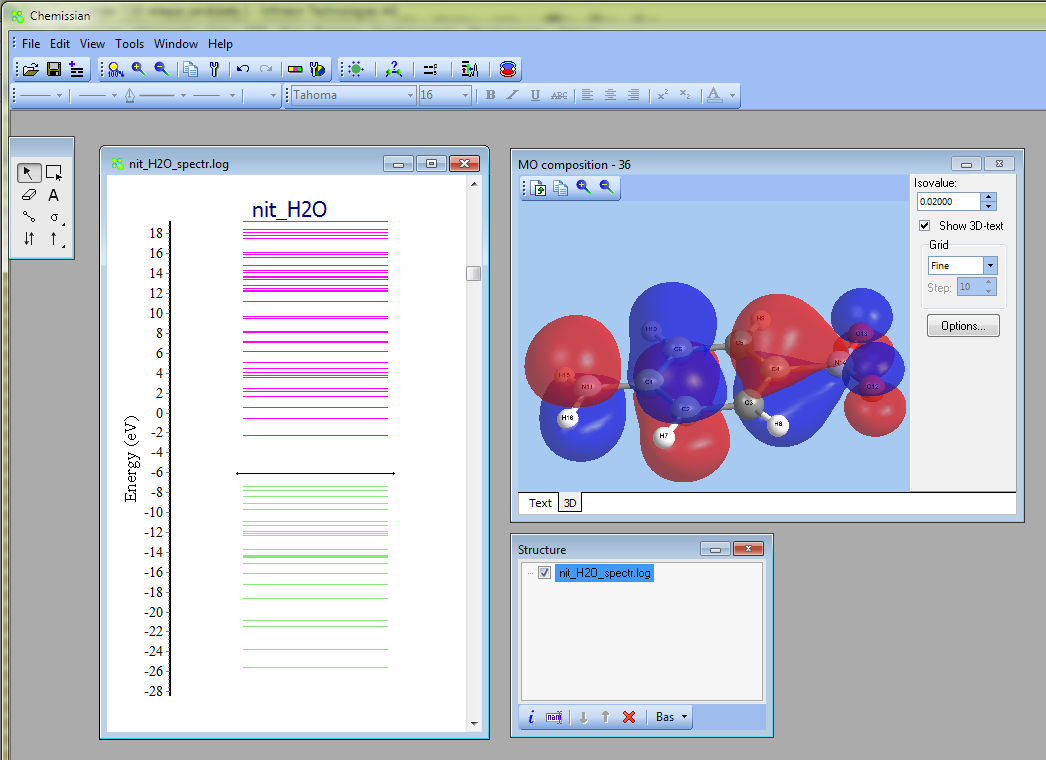
- NEW Support of Molpro output files via molden format
- UPDATEDImproved
"Analyze electron density":
- added support of 3D surfaces:
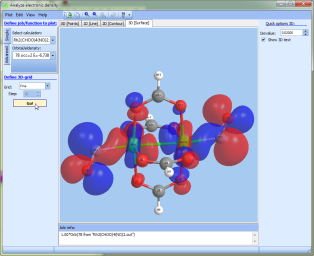
- in the 2D-mode (contour map) it is now possible to preview the plane in the frame of the 3D molecule.
- there is now predefined black and white color scheme for deformation density contour maps
- new options
- added support of 3D surfaces:
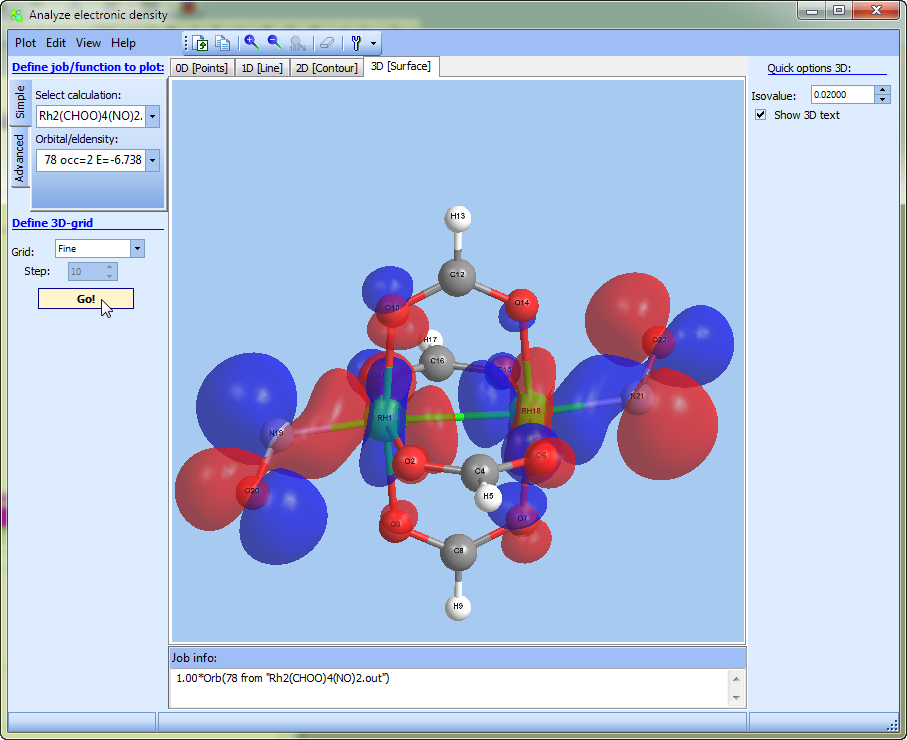
- Molecular orbitals calculated by Gamess program are now automatically displayed in pure form instead of Cartesian.
- UPDATEDImproved support of some kinds of Gaussian and Gamess files
[June 2011] Chemissian v2.2 released
- NEW Added support of EOM-CCSD spectra from Gaussian output files:
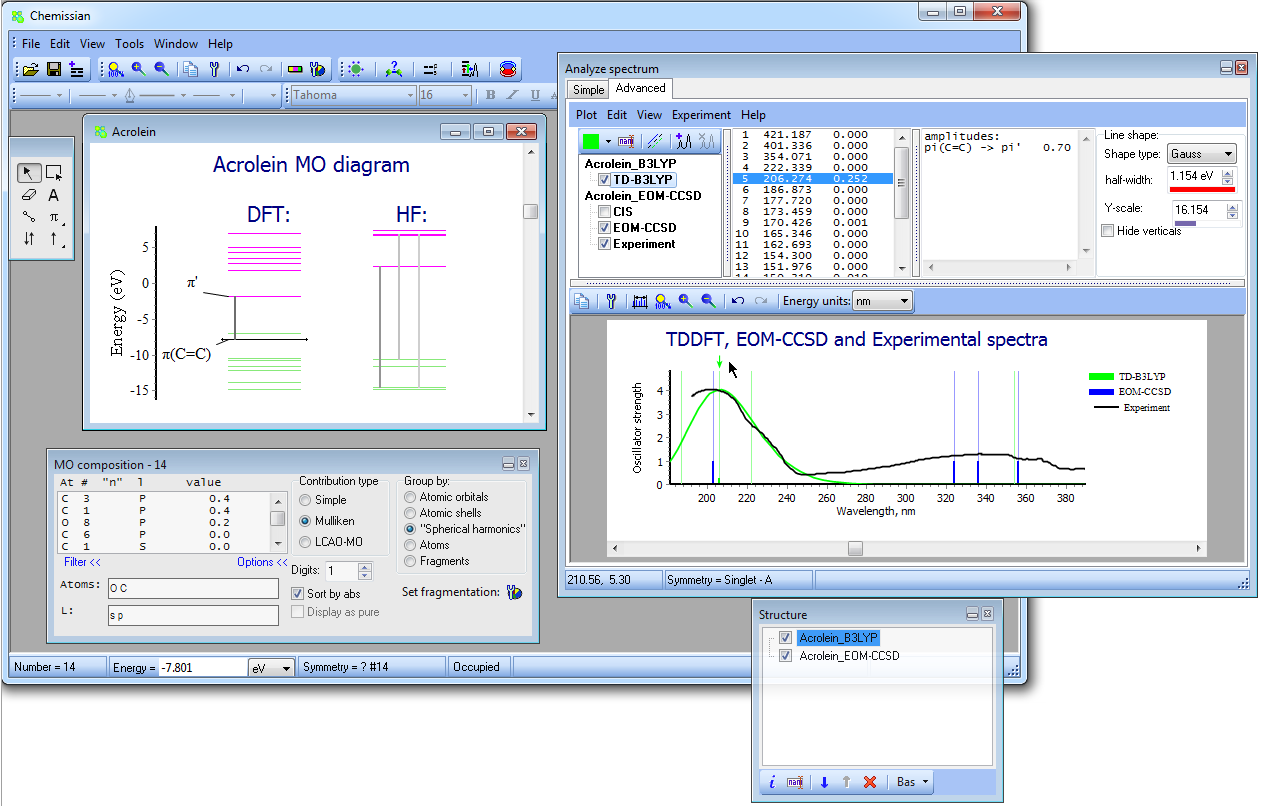{kind=link}
- NEW Added Energy shift option for calculated spectra, which is useful to obtain the total spectrum of excited states generated from different references (for more details see here).
- NEW Added export of basis set (and geometry) to GAMESS/Gaussian and Q-Chem input formats (see structure window)
- Fixed some bugs with reading Gaussian output files containing 6-31G* basis set.
[January 2011] Chemissian v2.0 released
- NEW Support of Q-Chem output files and support of Spartan output files are added. All features (visualization of molecular orbital diagrams, UV-VIS spectrum, electron density map, calculation of populations, quantum-chemical bond order indexes, etc.) are available. Some notes to the outputs can be found in online help.
- NEW Calculation of charge/spin density, MOs and any linear combination of them can be performed in any user-specified points. For details see "0D" section here.
- Other small improvements.
[January 2011]
Resolved problem with contactform[April 2010] Chemissian 1.771 released
- Fixed a bug occurring when reading g-functions from Gaussian: basis set was not correctly read, therefore density maps could be slightly inaccurate.
- UPDATED Improved "MO Composition" window:
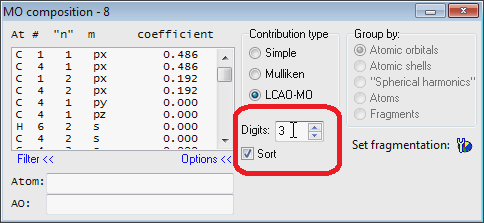
- UPDATED Now it is easier to associate text with selected MO level in the MO diagram editor: just press '.' on the keyboard and "Set MO Text" window will be shown and activated for text entering. Typed text will be automatically saved (there is no need to press the "Set" button).
[22 February 2010] Chemissian 1.77 released - what's new?
- UPDATED "AO Contribution in each MO Tool" --> "Analyze MO Compositions Tool":
- Now it is possible to consider contributions to the molecular orbitals by atomic orbitals, by atomic shells, by angular momentum ("spherical harmonics"), by atoms and by molecular fragments (e.g. groups of atoms).
- UPDATED "AO Coefficients window" -->"MO Composition window":
- This window displays the composition of the current MO in molecular orbitals energy level diagram editor. And now by clicking on the "option" button it is possible to configure how the contributions to the MO will be calculated (by AOs, atoms, fragments, etc. or just LCAO-MO coefficient (default)).
- UPDATED "Analyze populations and valences tool" -
- Now it is possible to group populations and valences by angular momentum.
[12 February 2010] Chemissian 1.751 released
Some problems when working in Windows Vista/Seven were solved.[9 February 2010] Chemissian 1.75 released - what's new?
- NEW Added "Analyze bonds tool":
Investigate bond order indexes and overlap populations between atoms and molecular fragments.
For more details see Analyze chemical bonding with Chemissian (and online help).
- "Analyze populations tool" --> "Analyze populations and valences tool":
NEWCalculate valence indexes for atoms, atomic orbitals, shells and molecular fragments.
For more details see Valences and Population analysis (and online help).
- UPDATED "Analyze electronic density tool" improvements (in 2D mode):
- Bonds between atoms are now shown.
- All atoms have individual sizes and colors.
- You can specify atoms that are to be shown: use the trackbar interface element near the "Show structure" checkbox to set the "cut" distance and it will be shown only those atoms whose distance to the plane is less than this "cut" distance. This option is useful when there are many atoms in a molecule and they hide the density map plane.
- Use mouse wheel to Zoom In / Zoom Out the contour density map plot and left mouse button to "drag" the plane.
For more details see online help
- Other small improvements